
We solve computational challenges in biomedical research that require statistical or algorithmic innovations. Much of our work is aimed at unlocking the full power of biobank-scale genetic data sets now becoming available (e.g., N=500,000 UK Biobank). To this end, we develop high-performance open source software resources freely available to the scientific community.
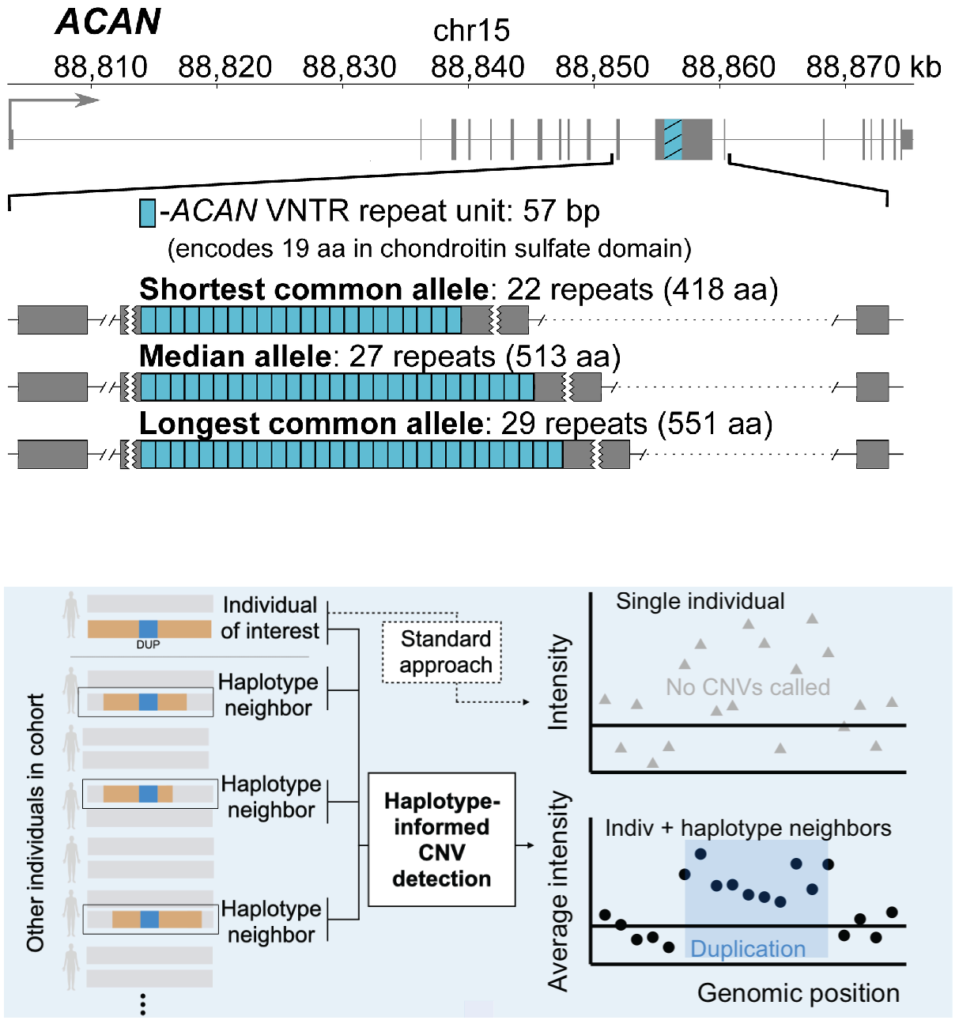
Haplotype-informed discovery of genetic variants shaping human phenotypes
Only a small subset of genetic variants known to associate with human traits involve polymorphisms that directly alter protein-coding sequence. We have recently undertaken several projects to expand this set of more-interpretable genetic associations by analyzing variation in parts of the coding genome previously inaccessible to biobank-scale analysis: namely, very rare coding variants and genomic structural variants. These analyses, made possible by new computational methods that leverage haplotype-sharing within biobank cohorts, have explained some of the strongest common-variant associations with human traits and identified many rare coding variants with large effect sizes (>0.5 standard deviations). We have also applied similar techniques to discover non-coding stuctural variants that strongly influence risk of common diseases.
Mukamel*, Handsaker* et al. medRxiv
Hujoel et al. 2022 Cell
Mukamel*, Handsaker* et al. 2021 Science
Barton et al. 2021 Nat Genet
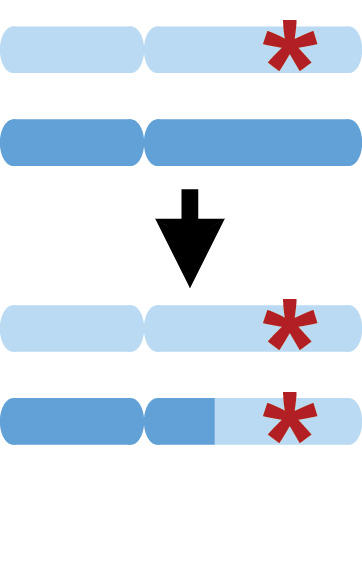
Mosaic chromosomal alterations
Somatic mutation is the process by which cells within an individual undergo DNA alterations, leading to “mosaic” mutations present in some (but not all) cells in the body. In some cases – most notably, cancers – mutant cells undergo clonal expansion, proliferating to high frequency. We have found that accurate statistical phasing enables highly sensitive detection of mosaic chromosomal alterations, revealing insights into the biology of clonal expansion and providing the potential for early detection of pre-cancerous mutations. We have also implicated mosaic chromosomal alterations (that likely arise early in development) as a likely contributor to sporadic cases of autism spectrum disorder.
Sherman et al. 2021 Nat Neurosci
Loh, Genovese, McCarroll 2020 Nature
Terao et al. 2020 Nature
Loh*, Genovese* et al. 2018 Nature
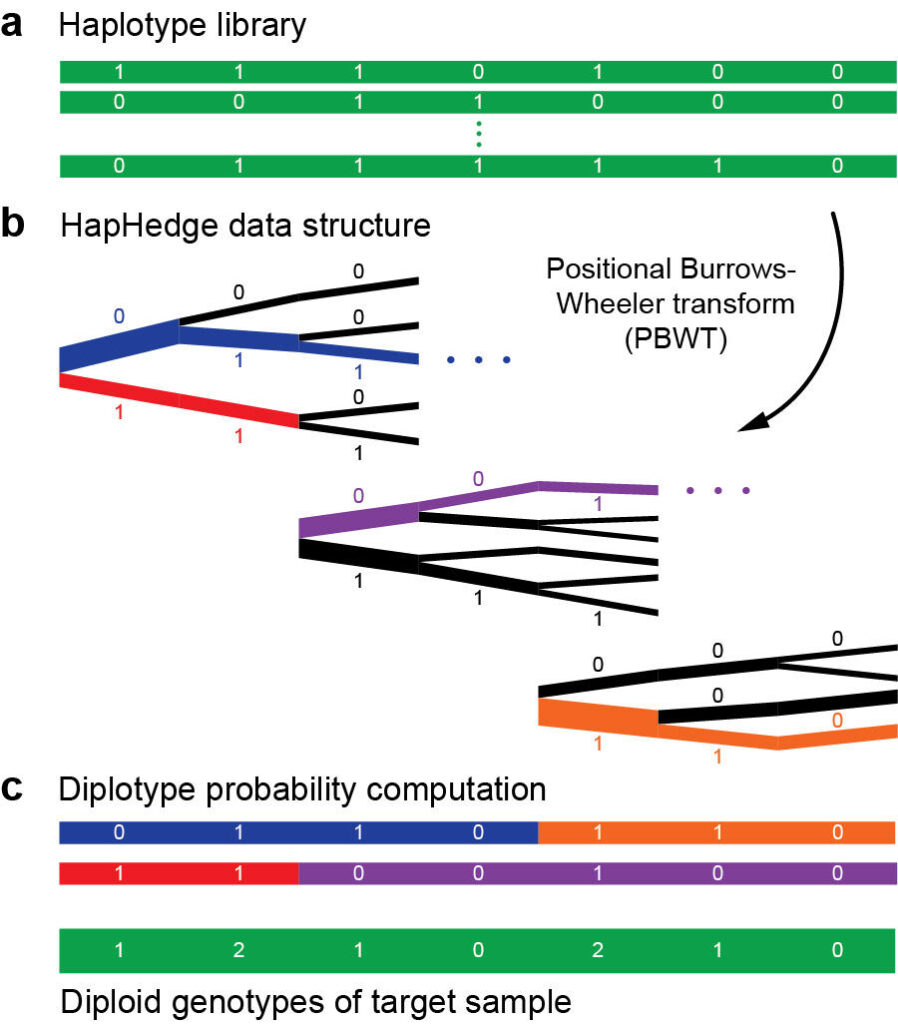
Haplotype phasing and imputation
The paradigm of phasing and imputation has emerged as the optimal means of maximizing statistical GWAS power within a given budget. In this paradigm, a large number of samples (the GWAS cohort) are genotyped at a small subset of genomic variants, and their genotypes at remaining variants are then statistically imputed using a whole-genome sequenced reference panel. This pipeline now enjoys near-universal use in GWAS, but as sample sizes have grown, computation has become a critical challenge – and opportunity for methodological innovation.
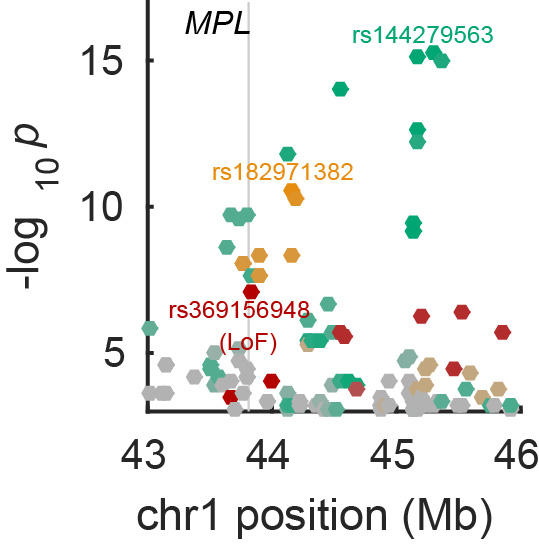
Genome-wide association analysis
Over the past decade, ever-larger genome-wide association studies (GWAS) have yielded rich insights into the genetic architectures of human complex traits. Linear mixed models (LMM) have become the method of choice for association analyses and have also proven versatile for partitioning heritability across the genome and performing phenotype prediction. We have developed and applied scalable LMM algorithms that detect and harness subtle statistical signals now becoming visible in very large data sets.
Loh et al. 2018 Nat Genet
Loh et al. 2015b Nat Genet
Loh et al. 2015a Nat Genet